Data Integrity
Understanding Data Integrity for
Environmental
Monitoring Equipment
by Jim Strachan, MBA
Climet Instruments Company
4 March 2019
Data integrity is an essential concept for regulated drug manufactures in today's digital age. In recent years, infractions related to data integrity have been noted in several Food and Drug Administration (FDA) warning letters (483's), import alerts, and consent decrees. Since 2017, Data Integrity has been a major audit concern for regulatory agencies in both North America and Europe.
Concerns about data integrity compliance has cause panic among many in the life science industry resulting in an over abundance of caution. Many managers as a result are falling into the trap of over-control, hence the colloquialism, "Death by Compliance." The main symptoms are the implementation of unnecessary controls, which either result in unnecessary costs, and/or an overabundance of procedures that virtually back producers into an audit corner. Worse, this over-control frequently has little to no impact on patient health. This is particularly true as it applies to environmental monitoring equipment.
This article reviews the primary sources for data integrity standards and guidance, discusses some of the more popular issues and concerns regarding particle counters, and provides industry best practices.
FDA 21 CFR Part 11
Title 21 of the FDA's Code of Federal Regulations (CFR) Part 11 (or 21 CFR Part 11) is the most widely used standard for appropriate data management. Part 11 applies to electronic format that are created, modified, maintained, archived, retrieved, or transmitted according to requirements set in FDA regulations. Electronic records/signatures that meet Part 11 requirements may be used in lieu of paper records. The standard (all sections combined) is approximately 8-9 pages long, and well worth a read.
The standard is divided into three parts:
| • |
General Provisions: |
|
| • |
Electronic Records:
|
|
| • | Electronic Signatures: |
ALCOA
ALCOA is the acronym used by the FDA that stands for Attributable, Legible, Contemporaneous, Original and Accurate.
It has been used by regulated industries as a framework for ensuring data integrity, and is key to Good Documentation Practice (GDP). The concept behind ALCOA is that data quality directly impacts product quality, with focus on performing tasks correctly the first time and immediate reporting of results. ALCOA is used in many regulatory documents, and it is important to be familiar with what is meant by each term (see above).


EU GMP Annex 1
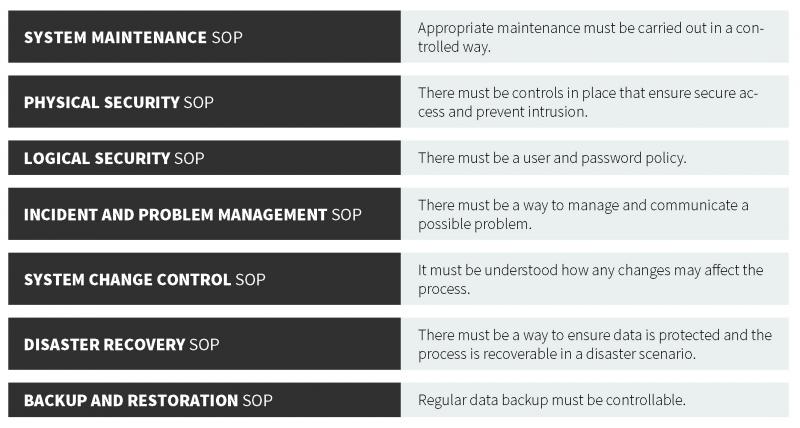
As part of the European Union (EU) EudraLex is a collection of rules and regulations governing medicinal products (for human and veterinary use). EU GMP Annex 1 (guidance, not a regulation), defines criteria for managing electronic records and signatures. These guidelines are similar to those of their U.S. counterpart. The central consideration of the EU GMP Annex 1 and 21 CFR Part 11 is to ensure that records are entered correctly, cannot be tampered with, can be stored for the retention period as well as retrieved (in full) at any time during use and during the retention period. In each regulation, there is a strong focus on record accuracy, integrity, security, and retrieval. From the guidance, there are several important components of a proper data management system, which forms the basis for the following recommended standard operating procedures (SOPs).
FDA Data Integrity and Compliance with Drug cGMP, Q&A, Guidance for Industry
This document provides guidance (i.e., is not a standard) and is at the time of this writing is in draft phase. It is intended to clarify the role of data integrity in current Good Manufacturing Practice (cGMP). The primary expectation is for data to be accurate and reliable. It does not establish legally enforceable responsibilities, but rather describes the FDA's current thinking. It is not a required document to adhere to, but is advisable to be familiar with the text. Key technical terms used in this document include: (1) Static record format, where static indicates a fixed data document such as a paper record or electronic signature; (2) Dynamic record format, which means a record that allows interaction between the user and record content, such as entering values manually in the system database; and (3) For a computer or related system, the "System" refers to the ANSI definition, which includes people, machines, and methods organized to accomplish a set of specific functions. In addition, this system can include computer hardware, software, peripherals, networks, cloud infrastructure, operators, and documents (manuals, SOPs, etc.).
The FDA also has published a Guidance for Industry, Part 11, which provide many insights.
MHRA GxP data integrity guidance
The (Medicines and Health products Regulatory Agency) MHRA GxP is a guidance document produced by the United Kingdom emphasizes that data integrity is fundamental to ensuring that medicines are of the required quality in a pharmaceutical quality system. The result of effective, robust data governance is complete, consistent, and accurate data used throughout the system. It primarily focuses on the importance of SOPs.
Company Culture and Ethics
It is clear from the FDA's Guidance for industry (and audit findings) that a company must have a quality culture. You can download the FDA's presentation by clicking here, or go to https://www.fda.gov/media/156357/download
Data integrity is a significant component of the Quality Management System, and inspectors around the world have made it very clear that good intentions are no defense against compromised data. The pharmaceutical industry must strongly consider any preventative or corrective action to improve product quality through an honest, ethical approach to data collection and retention. To safeguard against unethical behavior, the following are recommended industry best practices:
- • Particle Counter User Integrity:
Security MUST be enabled on all your portable particle counters. Each user must have a unique username and
password combination. Do not share login credentials. Maintain
a database of all user names, UserID's, passwords, and last
time password was changed (recommend annually).
- • Particle Counter Data Integrity: It is highly recommended that particle
counter sample data be automatically uploaded (via
Ethernet) into a LIMS or DAS database with little to no
human intervention, and retained on a Closed
System. That is, a secure network directory with limited
access, for example, restricted to the IT Department.
- • Enable Calibration Lock-out: Among the TOP 10 regulatory citations
is the use of equipment with an expired calibration.
- • When sample data is not directly uploaded from the
counter to a LIMS
or FMS database (i.e., from a USB Port), the files should be validated to ensure no
manipulation of data.
- • Data records should never be manipulated,
discarded, or destroyed.
- • Eliminate potential conflicts of interest - Limit access
to sample data files on the network or other storage devices
to the IT Department.
- • All records should be created automatically (and not created after the fact from memory).
- • Data should retain its unchanged time information.
- • All pertinent data should be included in the record (even if it is undesirable).
- • Passwords should never be shared between personnel.
- • All original records should be stored and kept safely on non-volatile media.
- • Everyone involved in the process, no matter their position, is responsible for upholding data integrity.
Test your knowledge
Given the focus of regulatory audits on data and user integrity, there has been in a frenzy among many in trying to interpret 21 CFR Part 11: Electronic Records, Electronic Signatures. Indeed, many biopharmaceutical and medical device producers are metaphorically, killing flies with sledgehammers by over interpreting 21 CFR Part 11.
Answer True or False to the following questions.
This is true
FDA's Data Integrity and Compliance with Drug CGMP (Section I, page 2), "It is the role of management with executive responsibility to create a quality culture where employees understand that data integrity is an organizational core confirms an increased emphasis on the critical role of senior management in creating a quality culture that identifies and addresses data integrity risks.
This is a key concept for the FDA.
This is true
FDA's Data Integrity and Compliance with Drug CGMP (Section III, No. 4, page 7) CGMP records or input of laboratory data into computerized records can be made by only authorized personnel (§211.68(b)). FDA recommends that manufacturers restrict the ability to alter specifications, process parameters, sample data, or manufacturing and test methods by technical means where possible (e.g., by limiting permissions to change settings or data).
The system administrator role, including any rights to alter files and settings, should be assigned to personnel independent from those responsible for the record content. To assist in controlling access, it is important that manufacturers establish and implement a method (a list) for documenting authorized personnel's access privileges for each CGMP computer system in use (§211.68(b)).
Industry best practice is that sample data from a particle counter should only be handled by system administrators.
This is a key concept for the FDA.
This is false
Per FDA's Guidance For Industry, Part 11, Section 3(b)(1), the FDA interprets 21 CFR Part 11 narrowly. In fact, this document mentions a narrow interpretation six (6) times.
This is true
Per 21 CFR Part 11, Section 11.10(d), the term "system" used in this clause applies to a Closed System. 21 CFR Part 11.3(b)(4) defines a Closed System as, "An environment in which system access is controlled by persons who are responsible for the content of electronic records that are on the system."
Section 11.10(d) states, that a particle counter, "Limits system access to authorized individuals."
21 CFR Part 11 does not require a specific number of security levels. However, from a practical stand point, more security levels allows for better customization and flexibility in implementing a security policy.
Climet CI-x5x Series of portable particle counters have four security levels with user authentication. Level 1 (User can collect data only), Level 2 (User can print reports and clear stored data), Level 3 (User can load internal programs), and Level 4 (Supervisor, free access to all menus and functions).
The Climet CI-x70 Series have five levels of security: Level 1 (User can collect data only), Level 2 (User can print reports and clear stored data), Level 3 (change program settings),Level 4 (access most menus and functions), and Level 5 (Supervisor, free access to all menus and functions).
Remote and continuous monitoring particle counters are controlled by software, and security is a function of end-user system administrator permissions.
This is true
Okay, this is sort of a trick question. 21 CFR Part 11.200(a) requires that login credentials not based on biometrics consist of two distinct identification components (i.e., a username and password), and that these credentials: (1) be unique so that no two individuals have the same combination; and (2) controls must, "Ensure that identification code and password issuance are periodically checked, recalled, or revised (e.g., to cover such events as password aging). Regarding the term "recall" or retirement of passwords mentioned in the previous sentence, the FDA would expect that upon the departure of an employee, the assigned electronic signature would be "retired" to prevent the former employee from falsely using the signature.
This is false
There is absolutely nothing in 21 CFR Part 11 (or elsewhere) that discusses password attempts or lockout.
This is false
There is nothing in 21 CFR Part 11 (or elsewhere) that discusses password complexity (i.e., number of digits, special characters, combination of lowercase and uppercase letters, and numbers). This is left up to the end-user and would be mentioned in their SOP.
Caution is advised here, that when writing an SOP it be done in a manner not to make it so restrictive as to be difficult to monitor or enforce.
This is false
There is nothing in 21 CFR Part 11 (or elsewhere) that requires passwords be changed at any specific frequency or interval.
21CFR11, Section 11.300(b) says that username and password issuances must be periodically checked, recalled, or revised (e.g., to cover such events as password aging).
Industry best practice is to update to login credentials and security settings on particle counters "at least" once a year, and this is generally conducted before, during, or after the annual plant shutdown. This must be addressed in an SOP.
Enforcing electronic signature expiration policies on individual particle counters can be ominous and tedious if you have multiple counters with multiple analysts. To confirm, it is not required by FDA 21 CFR Part 11, and is a perfect example of an over-interpretation (really a misunderstanding) of the 21 CFR Part 11 standard, which leads to unnecessary costs that have no patient benefit.
This is true
21 CFR §11.200(a)(2): Electronic signatures (a distinct identification code or "username" and a password ) shall be used only by their genuine owners and may never be shared.
This is false
There is nothing in 21 CFR Part 11 or elsewhere that prohibits changing data.
21CFR11, section 11.10(e) states that record changes shall not obscure previously recorded information. Such audit trail documentation shall be retained for a period at least as long as that required for the subject electronic records and shall be available for FDA review.
Industry best practice is to never change particle counter sample data.
This is false
Once more, there is nothing in 21 CFR Part 11 (or elsewhere) that requires sample data to be impervious to being changed or altered in an unauthorized fashion or manner (i.e., to be fraud proof). That said, Climet does use best efforts to ensure exported sample data from a particle counter is secure and cannot be altered, or otherwise validated. Furthermore, Climet offers the highest level of network and cyber security available to eliminate or substantially mitigate unauthorized network access.
Electronic data inside the Climet particle counter cannot be altered or changed, as it resides in a Closed System. However, after the sample data has been exported from the particle counter, there is always a way to change the data if someone is adequately motivated. For example, Portable Document Format (or PDF) files can be altered and re-saved. Even AES 256-bit military grade encryption can be cracked if given enough time and resources. In short, there is no way electronic data can be 100% secured.
This question more applies to a culture for honesty, integrity, and ethics (Refer to Question #1 above). Additionally, Standard Operating Procedures (SOPs) should be written to avoid or eliminate all potential conflicts of interest, and address what your organization is doing with the sample data to eliminate or substantially mitigate unauthorized changes (also known as fraud).
Industry best practice is that a System Administrator from the IT department who is not responsible for cleanroom quality be charged with exporting sample data and importing to a LIMS system. Also, it is recommended that sample data be uploaded to a LIMS automatically, without human intervention, on preferably a Closed System.
For example, Quality Analysts who take the sample data or other employees who are responsible for the air quality of a cleanroom, such as the Quality Control Manager or a Quality Assurance Manager, should not be involved in exporting sample data from a particle counter into a LIMS system or other database. This instead should be addressed by a procedure to eliminate any potential conflicts of interest.
Processes, procedures, training, and a culture of ethics are the real backbone of data integrity. The raw data itself is secondary
This is false
This is a very common misconception of 21 CFR Part 11. No where in this standard (or elsewhere) does it say that data, even more specifically particle counter sample data, must be in a non-editable format, or resistant to unauthorized changes.
However, I think it fair to say most end-user organizations and particle counter manufacturers (including Climet) have adopted a policy that requires particle counter data to be secured, and resistant to unauthorized modification or deletion under normal situations and conditions.
21CFR11, Section 11.10 preamble and subsection (e) refers to secure, computer-generated, time-stamped audit trails. It is most appropriate to mention that electronic data is most secure when processes and procedures (i.e., SOPs) are sufficient to protect unauthorized change or deletion of data. Refer to question #2 above.
This is true
A User Audit Trail is a particular feature of some portable particle counters that provide a pdf report (or similar) on who, when, and what changes in setup have occurred, and the individual or employee of each prior use of a particle counter. A User Audit Trail provides virtually worthless information from a data integrity perspective, and is not required by 21 CFR Part 11 (or elsewhere). Certainly, this level of reporting is not required for microbial air samplers, photometers, and other measurement instruments.
So, what does 21CFR11 require?
According to FDA Data Integrity and Compliance with Drug CGMP, Section II, sampling activities MUST be attributable to a specific individual (�211.100 and 211.160). This information (i.e., the UserID) is incorporated in each individual sample data file a Climet particle counter generates, and therefore each sample individually complies with FDA 21 CFR Part 11.
Additionally, to ensure satisfactory audit performance, industry best practices are to:
(1) Enable security and permissions on all portable particle counters, and only the ADMIN security level (limited to just a few senior managers) has permission to alter settings, delete sample data, etc.; The lowest level is only able to run samples, and cannot change or delete settings, samples, etc.
(2) Maintain User Integrity - - Each user must have a unique username and password combination and cannot be reused or re-assigned (i.e., do not shared) by anyone else;
(3) Maintain a log (for example, in a database, Microsoft Word, Excel, etc.) of all employees who are authorized to use or manage particle counters, list their security levels, and the last date the login credentials were checked, recalled, or revised;
(4) Periodic Review - - At least annually ensure login credentials on particle counters are checked, recalled, or revised (e.g., to cover such events as password aging); and
(5) When an employee is terminated, delete their login credentials immediately.
All of the aforementioned must be addressed in an SOP.
The Climet CI-x5x family of portable particle counters has four levels of security, and the CI-x70 Series has five levels of security. If you would like definitions for each security level, this is provided in the User Guide. Otherwise, please contact sales@climet.com.
The FDA's Guidance for Industry, Part 11 Section 3(B) confirms the FDA takes a narrow interpretation of 21CFR11, and goes on to say that broad interpretations of 21CFR11 (i.e., such as mandating a User Audit Trail on a particle counter), could lead to unnecessary controls and costs.
To better manage User Integrity, Climet recommends our PC Counter Cloning Utility, which helps to manage users, security levels, and settings on multiple CI-x5x particle counters. The CI-x70 Family have a cloning feature that makes management a snap.
If you would like a User Audit Trail feature for some other reason, it is available on our CI-x70 Series of portable particle counters.
This is false
A Climet particle counter is probably most closely associated with a network appliance. Sample data files resident in a Climet particle counter are created by the instrument itself. There is no mechanism to allow sample data to be modified. Network commands that can be input into a Climet particle counter are limited to properly formatted ASCII commands, and certainty there is no mechanism to save external image and other files (potentially infected with a virus or malware) into the particle counter.
No. A Climet particle counter does not need virus protection software per the reasons mentioned herein, and does not obtain operating system updates.
This is false
Actually, when only paper records are used, 21 CFR Part 11 does not apply (per Section 11.1(b)).
Referring to FDA's Guidance for Industry, Part 11 (lines 164-171), the short answer is there is nothing in 21 CFR Part 11 that prohibits the use of paper records. The problem really arises in how these paper records are handled. For example, are they hand typed into a LIMS system or database? This could potentially be a problem given human-error, which would negatively affect data integrity.
This is false
21 CFR Part 11 requires that no sample data be permanently deleted. Regulated users must: (1) document the reason why a specific sample should not be included; and (2) obtain necessary approval(s) per your SOP. The altered or aborted sample data record MUST have the same retention period as all other sample data records.
This is false
Included with the sale of each Climet particle counter that has a USB port, comes a USB Translator Utility. This software utility verifies the integrity of the data on the USB flash drive. Specifically, that the record contents have not been altered or corrupted (via checksum). If the sample data record is good the utility will push the record automatically to the secure network location.
This procedure and method fully complies with 21 CFR Part 11. In order to eliminate the potential for conflict of interest, it is highly recommended that by SOP someone not responsible for the sample data content be responsible for validation of the USB flash drive and uploading of the sample data to the LIMS.
This is true
To confirm, data integrity has more to do with a manufacturer's processes, procedures, employee training and corporate culture of ethics than it does with the actual data file itself. Additionally, there are also numerous ways to which sample data can be exported making it virtually impractical to do an IQ/OQ for data integrity.
Therefore, it is the end-user's responsibility to validate data integrity, mitigate or eliminate conflicts of interest, and write SOPs that comply with 21CFR11.
Companies interested in validating that sample data is being transmitted and received can simply run ten separate one minute samples, print the sample data on the particle counter, and validate the particle counts on the thermal print-out are displayed accurately in the database, LIMS system, or network location.
Again, how a company configures their network, and how they write their SOPs is the ultimately responsibility of the drug manufacturer.
This is false
This is an an actual comment made by an end-user, and a perfect example of a broad interpretation. Indeed, 21 CFR Part 11, §11.10(d) states that system access must be limited to authorized individuals. However, there is nothing in 21 CFR Part 11, or elsewhere, that explicitly states that date and timestamp must be an ADMIN function. On Climet's CI-x70 Series, the date and timestamp can be set by Access Levels 4 and 5. If an end-user only wants ADMINs to set the date and time, then simply do not assign anyone to Level 4.
In Conclusion
The best way of ensuring data integrity is to have a Closed System, which is defined by 21 CFR Part 11 as limited access to sample data files downloaded from a particle counter to a very small number of senior level employees. Moreover, Climet recommends that sample data be directly input into the LIMS database without human intervention. This can be accomplished by Climet's DataPro Software Suite. Virtually all LIMS systems can also pull Climet sample data files into its database directly from the particle counter via a script. Finally, some models of Climet particle counters can push sample data files to the network automatically, and when this is performed we recommend the directory location of the "pushed" data be secured with limited access by network security policies (i.e., a Closed System). And, that sample data be uploaded to the FMS by an employee who is not responsible for the sample data content.
Sources
FDA 21 CFR Part 11 or FDA website
FDA Guidance for Industry, Part 11
FDA Data Integrity and Compliance with Drug CGMP (Dec 2018)
MHRH 'GXP' Data Integrity Guidance and Definitions (May 2018)
This article is for information only, and may contain type-o's, errors, and omissions. Best efforts were made to ensure contents and citations were based on current sources, and there is no guarantee this article will be updated to reflect future changes to the standard or guidance. If you have specific questions we highly recommend contacting your Quality Assurance Manager, a consultant, or other competent expert.